Binding Chemistry
Binding Chemistry
Binding Chemistry
Chemical interactions tools and models helping you simulate and understand your molecule's binding behavior.
Chemical interactions tools and models helping you simulate and understand your molecule's binding behavior.
Achieve unified intelligence
Equipped with Models for Every Challenge
Equipped with Models for Every Challenge

Static Biochemical Interactions
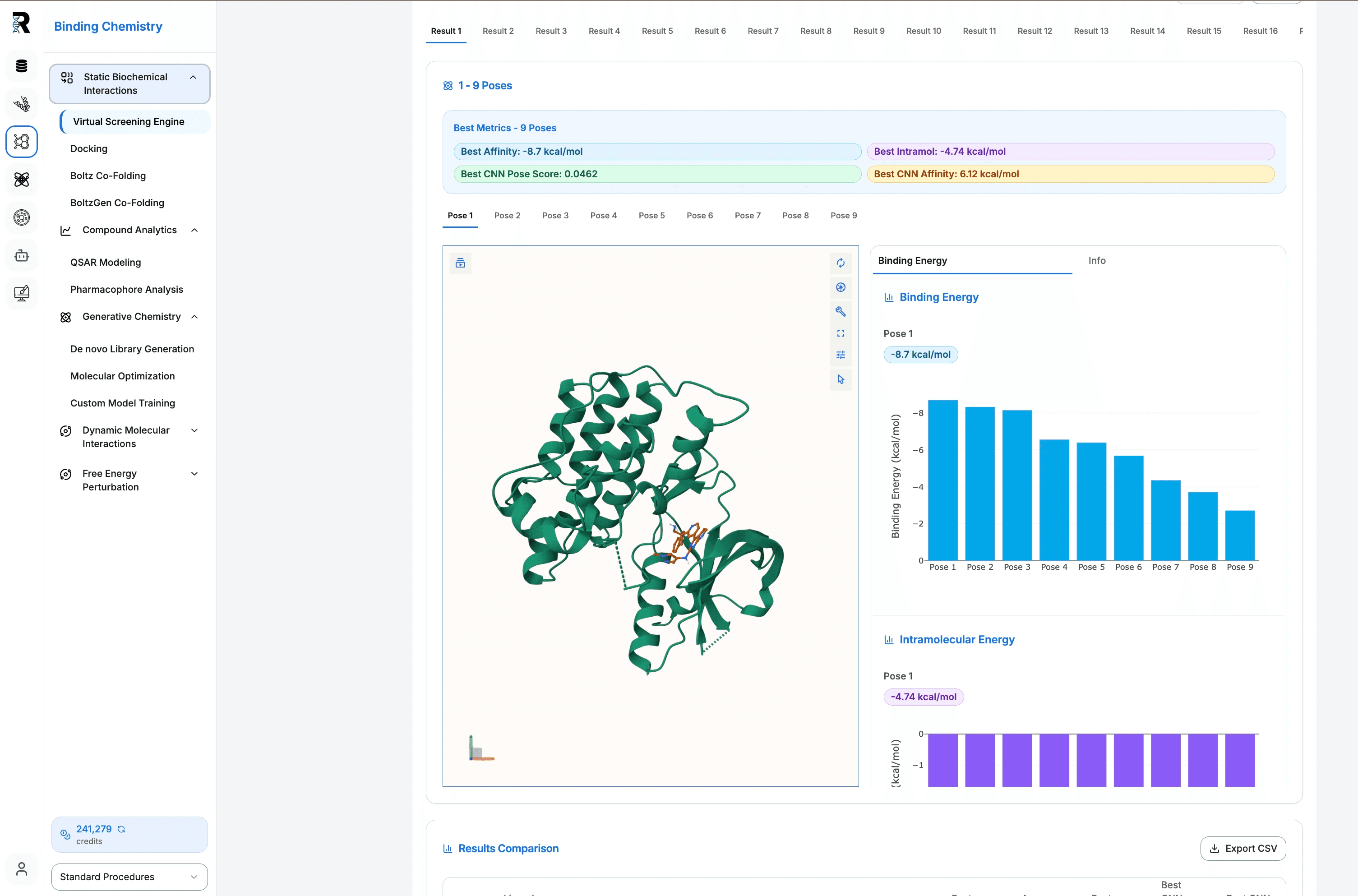
Virtual Screening
Screen your molecular libraries using high-throughput static, flexible, and ensemble docking with seamless binding affinity and conformational analysis.

Static Biochemical Interactions
Boltz Co-Folding
Test binding affinity metrics between your molecule and associated target proteins for precise biomolecular interaction predictions.

Static Biochemical Interactions
BoltzGen Co-Folding
Generate binding models between your designed molecules, peptides, or proteins, and target protein structures across multiple interaction pairings.

Compound Analytics
QSAR Modeling
Upload molecules and their associated data to view cluster-level statistics and perform substructure/motif analysis.

Compound Analytics
Pharmacophore Analysis
Analyze protein-ligand interactions for multiple complexes with atomistic 2D interaction maps and 3D visualization.

Generative Chemistry
Generative Chemistry
Generate novel chemistries using a variety of reinforcement learning models, expand your lead series, and run multi-parameter optimization on your compounds.

Dynamic Molecular Interactions
Protein Water MD
Simulate desired proteins in water across various conditions such as ion and solvent parameters, energies, temperatures, and time scales.

Dynamic Molecular Interactions
Protein Ligand
Simulate protein–ligand binding with high fidelity dynamics in realistic solvent and ionic environments over extended timescales.

Dynamic Molecular Interactions
Ligand Membrane
Simulate compound permeability across 100+ membrane types under realistic dynamic pulling conditions.

Free Energy Perturbation
ABFE/RBFE Engine
Calculate absolute or relative binding free energy for your ligand-protein pairs with select protein anchors, force fields, and simulation parameters.

Free Energy Perturbation
Absolute Binding FE
Compute absolute binding free energies using alchemical FEP with full molecular mechanics, force‑field precision, and integrated ligand–protein systems.
Static Biochemical Interactions
Virtual Screening
Screen your molecular libraries using high-throughput static, flexible, and ensemble docking with seamless binding affinity and conformational analysis.
Static Biochemical Interactions
Boltz Co-Folding
Test binding affinity metrics between your molecule and associated target proteins for precise biomolecular interaction predictions.
Static Biochemical Interactions
BoltzGen Co-Folding
Generate binding models between your designed molecules, peptides, or proteins, and target protein structures across multiple interaction pairings.
Compound Analytics
QSAR Modeling
Upload molecules and their associated data to view cluster-level statistics and perform substructure/motif analysis.
Compound Analytics
Pharmacophore Analysis
Analyze protein-ligand interactions for multiple complexes with atomistic 2D interaction maps and 3D visualization.
Generative Chemistry
Generative Chemistry
Generate novel chemistries using a variety of reinforcement learning models, expand your lead series, and run multi-parameter optimization on your compounds.
Dynamic Molecular Interactions
Protein Water MD
Simulate desired proteins in water across various conditions such as ion and solvent parameters, energies, temperatures, and time scales.
Dynamic Molecular Interactions
Protein Ligand MD
Simulate protein–ligand binding with high fidelity dynamics in realistic solvent and ionic environments over extended timescales.
Dynamic Molecular Interactions
Ligand Membrane MD
Simulate compound permeability across 100+ membrane types under realistic dynamic pulling conditions.
Free Energy Perturbation
ABFE/RBFE Engine
Calculate absolute or relative binding free energy for your ligand-protein pairs with select protein anchors, force fields, and simulation parameters.
Free Energy Perturbation
Absolute Binding FE
Compute absolute binding free energies using alchemical FEP with full molecular mechanics, force‑field precision, and integrated ligand–protein systems.

The Final Operating System of Pharma.

The Final Operating System of Pharma.